Sindromul Crouzon, de asemenea Boala lui Crouzon numit, este una dintre mai multe craniosinostozele cauzate genetic, în care suturile craniene se osifică prematur, astfel încât creșterea craniului este perturbată și pot apărea malformații și aderențe tipice pe cap și față. Dezvoltarea mentală a persoanelor afectate de sindromul Crouzon este de obicei normală.
Care este sindromul Crouzon?

© royaltystockphoto - stock.adobe.com
De asemenea, sindromul Crouzon Disostoză craniofacială Crouzon numit, este unul dintre mai multe craniosinostozele cunoscute. Boala se caracterizează prin osificarea precoce a suturilor craniene, unele începând prenatal. Osificarea înseamnă că, în faza de creștere, creierul nu se poate extinde ușor sub craniu, care în mod normal „crește cu copilul”. În schimb, craniul crește în principal pe suturile craniene încă nu osificate, astfel încât, dacă sunt lăsate netratate, apar malformații tipice.
$config[ads_text1] not found
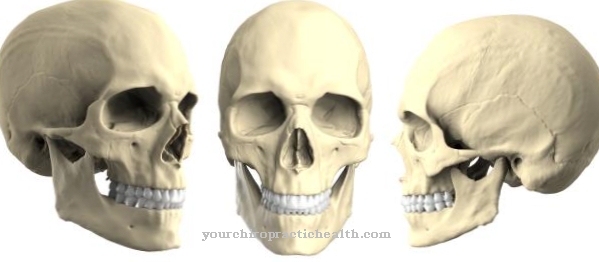
În sindromul Crouzon, suturile coronare, alfa și sagitale se osifică inițial. Fără tratament sau fără intervenții chirurgicale corective, apare un craniu turn tipic și anomalii faciale, cum ar fi ușurarea excesivă a ochilor (hipertelorism) și ochii proeminenți (exoftalmos). În plus față de alinierea necorespunzătoare a dinților, în sindromul Crouzon trebuie să se aștepte surditatea, deoarece canalul auditiv extern are ocluzie, un atrezie canal auditiv și / sau osiculele nu sunt pe deplin dezvoltate, astfel încât apare deficiența de auz.
cauze
Sindromul Crouzon este cauzat exclusiv de o mutație a locusului genei 10q26 de pe cromozomul 10. Pe cromozomul 10 există aproximativ 1.200 de gene, care conțin de la 4% la 4,5% din ADN-ul celulelor umane. Gena 10q26 este responsabilă de codificarea „receptorului 2 al factorului de creștere a fibroblastului” (FGFR2). Efectele acestei mutații specifice genelor sunt pronunțate diferit într-un interval observat.
$config[ads_text2] not foundMutația genelor este moștenită ca o trăsătură dominantă autosomală. Acest lucru înseamnă că sindromul Crouzon nu este specific sexului, deci poate afecta bărbații și femeile în egală măsură și înseamnă că boala va apărea cu siguranță chiar dacă doar un părinte este afectat de defectul genetic de la locus 10q26. Cel mai vizibil efect al acestui defect genetic este osificarea prematură a suturilor craniului. Suturile craniene reprezintă plăcile de creștere ale plăcilor osoase ale osului frontal (Os frontale), osului parietal (Os parietale) și osului occipital (Os occipitale).
Dacă suturile se osifică în timpul fazei de creștere, craniul nu se poate lărgi uniform și creierul provoacă creșterea presiunii de creștere, ceea ce duce la deformările tipice ale craniului.
Simptome, afectiuni si semne
În plus față de simptomele deosebit de vizibile ale sindromului Crouzon deja descrise mai sus, cum ar fi cranii turn, ochi proeminenți și relief larg al ochilor, există și alte semne care indică prezența sindromului Crouzon. Acestea sunt suturile craniene proaspete osificate, poziția slabă a ochilor și strabismul. Strabismul este o lipsă de coordonare a mușchilor oculari.
Ochii nu pot fi paralelați sau aliniați la un obiect comun. Hipoplazia maxilarului superior și o buză proeminentă inferioară sunt, de asemenea, efecte secundare ale sindromului Crouzon. Simptomatica hipoplaziei maxilare, cunoscută și sub denumirea de retrognatie maxilară, este bărbia care iese departe de maxilă.
$config[ads_text3] not found
În general, rezultatul este imaginea unei expresii faciale concave. De regulă, manifestările simptomatice ale sindromului Crouzon nu se limitează doar la craniu, ci apar alte probleme „asociate”. Trebuie menționată sinostoza humero-radială, o osificare parțială a articulației umărului și o subluxare în articulația cotului.
Diagnostic și curs
O suspiciune cu privire la o posibilă prezență a sindromului Crouzon poate apărea prenatal pe baza istoricului familial. Simptomele vizibile extern fac imaginea de diagnostic aproape de prisos. Dacă există vreo îndoială cu privire la prezența sindromului Crouzon, o analiză genetică poate oferi informații. Cursul bolii variază de la o persoană la alta, în special în cazul imaginilor clinice asociate.
Dacă boala este lăsată netratată, simptomele principale tind să apară în timpul fazei principale de creștere a craniului și a creierului. După ce s-a încheiat faza de creștere, deformațiile vizibile clar ale capului și feței rămân pe tot parcursul vieții, cu excepția cazului în care sunt efectuate intervenții chirurgicale.
complicaţiile
De regulă, formarea craniului este afectată în special de sindromul Crouzon, ceea ce duce la osificarea suturilor craniului. Aceasta poate provoca deformări enorme pe cap, care afectează foarte mult aspectul pacientului. Majoritatea oamenilor suferă de scăderea stimei de sine și se simt neatractivi.
Sindromul Crouzon poate duce și la dificultăți sociale, ceea ce se întâmplă mai ales la copii și tineri. Aspectul schimbat poate duce la bullying. În sindromul Crouzon, dezvoltarea mentală nu este afectată în majoritatea cazurilor. Ochii sunt, de asemenea, afectați de sindromul Crouzon, astfel încât poate apărea un strabism. Acest lucru duce la dificultăți de coordonare.
$config[ads_text4] not foundComplicațiile apar atunci când sindromul Crouzon nu este tratat chirurgical în copilărie. Tratamentul în sine este posibil numai ca măsură chirurgicală și vizează în primul rând corectarea malformațiilor. Mai presus de toate, spațiul este creat pentru creierul în creștere. Cu toate acestea, o vindecare completă a sindromului nu este posibilă. Nu există nicio reducere a speranței de viață, atâta timp cât nu există complicații speciale în cursul operației.
Când trebuie să te duci la doctor?
În majoritatea cazurilor, sindromul Crouzon este recunoscut imediat după naștere sau chiar înainte de naștere, astfel încât, în majoritatea cazurilor, nu este necesar un diagnostic suplimentar. Cu toate acestea, trebuie consultat un medic pentru a trata plângerile și malformațiile individuale.
În special, în cazul în care copilul are un obraz, trebuie consultat un medic pentru a remedia această plângere. Mușchii feței pot fi afectați și de sindromul Crouzon, de aceea este necesară o vizită la medic dacă pacientul nu este în măsură să formeze o expresie facială independentă.
Orificarea articulațiilor poate indica, de asemenea, sindromul și trebuie examinat. De regulă, simptomele pot fi examinate și diagnosticate de către un medic pediatru sau un medic generalist. Tratamentul suplimentar depinde de severitatea simptomelor, astfel încât intervențiile chirurgicale pot fi necesare.
În cazul în care copilul sau rudele și părinții lor se confruntă cu reclamații psihologice din cauza sindromului Crouzon, trebuie consultat și un psiholog pentru a evita complicațiile și complicațiile suplimentare.
Medici și terapeuți din zona dvs.
Tratament și terapie
Tratamentul sindromului Crouzon constă în esență - dacă este deloc - dintr-o intervenție chirurgicală corectivă. Există trei tehnici chirurgicale diferite cunoscute care sunt oferite de clinici de specialitate. Avansul orbital frontal constă în tăierea frontului a craniului, inclusiv a frunții, în craniu și re-fixarea acestuia, astfel încât creierul să aibă loc pentru creșterea necesară.
Reasamblarea craniului se poate face practic cu ajutorul plăcilor de titan, cu un sistem de plăci absorbabile sau cu material de sutură absorbabil. Ce metodă este utilizată depinde de condițiile întâlnite în timpul operației. Operațiile asupra craniului facial osos sunt de obicei mai complexe și sunt cunoscute sub numele de osteotomia Le Fort III. În unele cazuri, acest lucru poate corecta și o poziție a ochilor prea largă.
A treia procedură, osteogeneza de distragere, permite deplasarea treptată a plăcilor craniului. Dispozitivele de distragere destinate anumitor zone ale craniului sunt utilizate chirurgical, iar la doar câteva zile după operație, plăcile osoase pot fi îndepărtate unele de altele cu până la un milimetru în fiecare zi folosind sistemul de fixare încorporat. Osul umple golul cu țesutul calus, care ulterior osifică, creând un fel de creștere artificială a craniului.
Perspective și prognoză
Sindromul Crouzon trebuie întotdeauna tratat. Dacă sindromul nu este tratat, duce de obicei la moarte. Tratamentul poate fi bazat doar pe simptomele sindromului și nu poate fi cauzal.
Malformațiile sunt corectate cu ajutorul intervențiilor chirurgicale. Diagnosticul precoce și tratamentul au un efect foarte pozitiv asupra evoluției ulterioare a bolii. O operație timpurie oferă creierului suficient spațiu pentru o creștere sănătoasă, astfel încât să nu mai existe restricții sau disconfort în viața pacientului.
De regulă, persoana în cauză nu suferă de alte simptome după procedură și nu există complicații. De asemenea, dezvoltarea mentală a bolnavului nu este perturbată de boală dacă este tratată din timp. Chiar și după un tratament de succes, examinările periodice sunt recomandate pentru a evita simptomele ulterioare.
De regulă, sindromul Crouzon nu are un impact negativ asupra speranței de viață a persoanei afectate dacă este descoperită precoce și tratată complet. Dacă sindromul Crouzon nu este tratat, va duce la malformații suplimentare la nivelul feței și, astfel, la restricții în viața persoanei afectate. Articulațiile umerilor și ochilor sunt afectate în special.
Împiedica
Deoarece apariția sindromului Crouzon este genetică, nu se cunosc măsuri preventive directe. Nu există măsuri care să poată preveni boala de la sine. Cu toate acestea, în cazul în care există o suspiciune a defectului genetic cauzal, măsurile preventive sunt importante pentru a reduce la minimum efectele bolii - în special în faza de creștere.
Măsurile preventive constau într-o inspecție vizuală regulată a craniului și verificarea presiunii intracraniene pentru a oferi creierului posibilitatea creșterii normale sub nivelul craniului cu ajutorul unei operații pe craniu.
Dupa ingrijire
Cu sindromul Crouzon, în majoritatea cazurilor nu există măsuri speciale de urmărire disponibile pentru persoanele afectate. Cu această boală, pacientul depinde în primul rând de un diagnostic rapid și de un tratament ulterior, deoarece acesta este singurul mod de a evita complicațiile suplimentare sau agravarea în continuare a simptomelor.
Prin urmare, trebuie consultat un medic la primele semne ale bolii. Tratamentul începe mai devreme, cu atât va fi mai bine cursul ulterior al acestei boli. Întrucât aceasta este o boală genetică, consilierea genetică trebuie întotdeauna efectuată în primul rând dacă doriți să aveți copii. Acest lucru poate preveni apariția din nou a sindromului Crouzon la descendenți.
Tratamentul se realizează printr-o procedură chirurgicală. Persoana afectată ar trebui să se odihnească definitiv după această procedură și să aibă grijă de corpul său. Ar trebui să vă abțineți de la efort sau de la activități fizice stresante și fizice pentru a nu împovăra inutil corpul. În plus, sprijinul oferit de propria familie sau de la prieteni și cunoscuți este adesea foarte important. În cele mai multe cazuri, această boală nu reduce speranța de viață a persoanei afectate.
Puteți face asta singur
Când un copil se naște cu sindromul Crouzon, părinții sunt întâi provocați. Este important ca pacientul să fie supus unui diagnostic cuprinzător. Pot fi create planuri de terapie care includ atât intervenții chirurgicale, cât și alegerea de ajutoare medicale. În special, operațiile craniului trebuie să fie efectuate cât mai devreme.
Cu toate acestea, pacienții cu sindromul Crouzon nu necesită doar tratament medical extins. De obicei, sunt priviți, marginalizați sau hărțuiți de la o vârstă fragedă. Puteți ajuta aici psihoterapia empatică, în care părinții și frații ar trebui să fie implicați.
Adesea este util ca părinții și pacienții să intre în contact cu alte persoane afectate. Există mai multe posibilități pentru aceasta.De exemplu, site-ul web „Parents’ Initiative Apert Syndrome and Related Malformations e.V. ”(www.apert-syndrom.de) oferă și informații despre sindromul Crouzon și oferă, de asemenea, un eveniment de formare anuală ca reuniune familială.
Aproximativ optzeci de pacienți cu sindromul Crouzon sunt înregistrați în prezent pe site-ul „Diseasemaps”. Dacă vă alăturați acestui site - de renume -, puteți contacta pacienții individuali din țările respective (www.diseasemaps.org/de/crouzon-syndrome).
Deoarece sindromul este moștenit în mod autosomal dominant, pacienții cu sindromul Crouzon ar trebui să solicite sfaturi genetice atunci când încearcă să conceapă.






.jpg)





.jpg)