Sindromul Holt-Oram este un sindrom de malformație care este în principal asociat cu defecte cardiace și anomalii ale degetelor și este cauzat de o mutație. În majoritatea cazurilor, mutația cauzativă are loc sporadic și corespunde astfel unei mutații noi. Corecția chirurgicală a defectului cardiac este punctul central al terapiei.
Sindromul Holt-Oram?

© SmirkDingo - stock.adobe.com
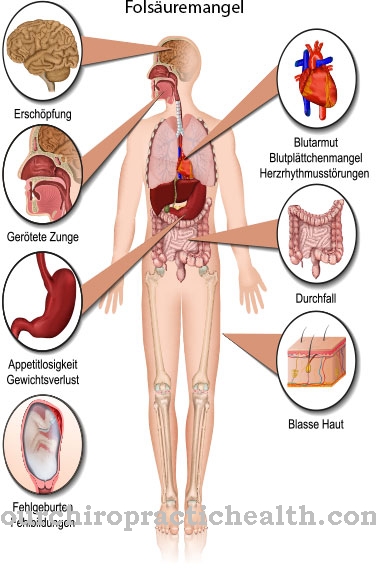
Sindroamele congenitale de malformație cu implicarea predominantă a extremităților sunt un grup de boli care include diverse deformări ale brațelor și picioarelor. Unul dintre ei este acesta Sindromul Holt-Oram, numit si Sindromul inimă-mână este cunoscut. Sindromul este asociat cu grupul de displazii atriodigitale, care includ boli congenitale cu malformații ale extremităților superioare și ale inimii.
$config[ads_text1] not found
Pe lângă sindromul Holt-Oram, sindromul inimă-mână tip 2, sindromul inimă-mână tip 3 și sindromul mâinii inimii de tip slovean aparțin acestui grup de boli. Sindromul Holt-Oram a fost descris pentru prima dată în 1960. Pediatrul britanic Holt și cardiologul Samuel Oram sunt primii care descriu boala. Sindromul Holt-Oram Aceasta este o boală relativ rară și este asociată cu o prevalență medie a unei persoane afectate la 100.000 de persoane.
Cauza malformațiilor mâinilor și inimii în sindromul Holt-Oram constă în genetică. Simptomele sindromului sunt la fel de variate ca și cauzele sale suspectate. Malformațiile din boală se concentrează asupra inimii și a degetului mare.
cauze
Deși sindromul Holt-Oram este o boală genetică și congenitală, nu există aproape o frecvență familială în cazurile documentate până acum. Deși sindromul pare a fi moștenit într-un mod autosomal dominant de moștenire în cazuri individuale, o mare parte din documentația cazului sugerează apariția sa sporadică. Aproximativ 85 la sută din cazurile documentate par să se datoreze unei mutații noi.
$config[ads_text2] not foundCauza principală a sindromului Holt-Oram constă în mutații genetice la nivelul locului genei 12q23-24.1 La această genă se află așa-numita genă TBX5, care este localizată pe cromozomul 12 și pentru o proteină implicată la membre și membre. Dezvoltare cardiacă codificată. Funcțiile exacte ale proteinei nu au fost încă clarificate. De asemenea, nu s-a stabilit încă dacă factorii externi precum expunerea la toxine sau malnutriția la mamă în timpul sarcinii favorizează mutația genei TBX5.
Mutațiile genei pot fi detectate la cel puțin 70 de persoane din 100 de pacienți cu sindrom Holt-Oram. Cu toate acestea, știința presupune că anomaliile din alte gene pot provoca, de asemenea, simptomele sindromului. De exemplu, sindromul de malformație este asociat cu polisindactia cu degetul tripartit.
Simptome, afectiuni si semne
Pacienții cu sindrom Holt-Osram suferă de un complex de malformații care afectează în principal degetul mare și inima. Deși localizarea malformațiilor pacientului este frecventă, sunt posibile diferite tipuri de malformații pe inimă și degetul mare. Prin urmare, tabloul clinic este extrem de variat.
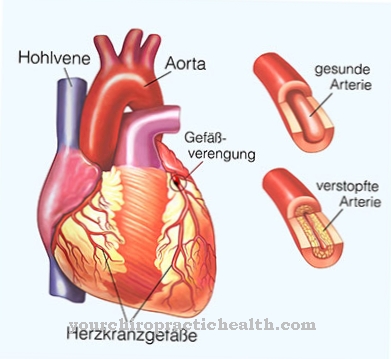
Defectele cardiace se pot manifesta, de exemplu, sub forma unui defect septal ventricular, un defect septal atrial, o aritmie cardiacă sau o tulburare de conducere. Anomaliile scheletului pot corespunde malformațiilor de reducere a degetelor, dar pot apărea și anomalii, cum ar fi neaplicarea vorbei.
$config[ads_text3] not foundMulți pacienți cu sindrom suferă, de asemenea, de sinostoze radioulnare și anomalii ale coastelor, scapulei sau claviculei. În plus, sindromul Holt-Oram este asociat cu pectus carinatum și scolioză. Majoritatea celor afectați suferă, de asemenea, de sindactilie a degetelor sau a falangelor de la nivelul picioarelor. În cazuri individuale aceste simptome sunt asociate cu hipertelorism.
Diagnosticul și cursul bolii
Sindromul Holt-Oram este adesea diagnosticat greșit. Într-un diagnostic diferențiat, medicul trebuie să diferențieze complexul de simptome de sindromul Okihiro după mutațiile genice din gena SALL4 pe cromozomul 20, care este asociat cu aceleași malformații ale brațului și defecte cardiace. Ceea ce este deosebit de relevant în diagnosticul diferențial este că pacienții cu sindrom Okihiro au de obicei o anomalie Duane.
Ei stârnesc, sunt adesea afectați de malformații renale și au tulburări de auz, anomalii ale piciorului sau malformații ale urechii. Sindromul Trombocitopenie-Radius-Absent-Radius trebuie, de asemenea, diferențiat de sindromul Holt-Oram, care se realizează în principal prin diagnostice de laborator. Alte imagini clinice cu o imagine similară clinic sunt anemia Fanconi și sindromul Pallister Hall.
Speranța de viață a pacienților cu sindrom Holt-Oram nu este sub medie. Doar în cazuri grave există un defect cardiac greu de tratat, care face ca prognosticul să fie nefavorabil.
complicaţiile
Sindromul Holt-Oram determină o serie de malformații și deformări diferite la pacient, ceea ce poate îngreuna viața și viața de zi cu zi. Mai presus de toate, inima este afectată de malformații, astfel încât pacientul suferă de un defect cardiac. Există, de asemenea, o aritmie cardiacă din care persoana afectată poate muri în cel mai rău caz.
$config[ads_text4] not foundAnomaliile apar și pe degetul mare, astfel încât anumite mișcări sau procese din viața de zi cu zi sunt de asemenea mai dificile. Nu este neobișnuit ca deformările de pe corp să conducă la tachinarea și intimidarea altor copii, ceea ce poate duce la plângeri psihologice și depresie la mulți pacienți. Nu este neobișnuit să apară afecțiuni renale, ceea ce în cel mai rău caz poate duce la insuficiență renală.
Mai mult, persoanele afectate suferă, de asemenea, de o deficiență vizuală și o deficiență de auz. De regulă, speranța de viață rămâne neschimbată ca urmare a sindromului Holt-Oram, atât timp cât nu există un defect cardiac care să conducă la moarte. Un tratament cauzal al sindromului Holt-Oram nu este de obicei posibil, astfel încât doar simptomele pot fi tratate. În multe cazuri, este necesar și sprijin psihologic.
Când trebuie să te duci la doctor?
Sindromul Holt-Oram este de obicei diagnosticat la scurt timp după naștere. În funcție de cât de grave sunt malformațiile, copilul afectat poate avea nevoie de examene medicale suplimentare. În principiu, defectele cardiace trebuie tratate prompt pentru a reduce riscul de boli secundare grave. Dacă apar complicații, cum ar fi aritmii cardiace sau semne de defect septal atrial, este necesar un sfat medical. De asemenea, trebuie consultat un profesionist medical cu anomalii ale degetului scheletic.
Părinții copiilor afectați ar trebui să consulte cu atenție medicul și să-i informeze despre orice simptome neobișnuite. Deoarece sindromul Holt-Oram este o boală ereditară, nu este posibil niciun tratament cauzal. Prin urmare, poate fi necesar ca pacienții să fie tratați o viață, în funcție de care apar malformații și care este constituția pacientului. Deoarece de multe ori acest lucru provoacă și plângeri emoționale, este indicat sprijinul psihologic însoțitor. Copiii care suferă de intimidare sau tachinare trebuie să solicite consiliere terapeutică cu părinții.
Medici și terapeuți din zona dvs.
Terapie și tratament
Nu sunt disponibile tratamente cauzale pentru pacienții cu sindrom Holt-Oram. Există speranță pentru o tratabilitate cauzală în viitor, deoarece terapia genică este în prezent obiectul cercetărilor medicale. Cu toate acestea, atâta timp cât acest tip de terapie nu ajunge în faza clinică, sindromul Holt-Oram rămâne o boală incurabilă.
În acest moment, sunt disponibile doar opțiuni de terapie simptomatică pentru tratamentul pacienților. Terapia se bazează pe simptomele cazului individual. Corecția precoce a defectului cardiac are o relevanță deosebită. Această corecție se face chirurgical. În cazul unui defect septal Vohof, procedura chirurgicală are ca scop închiderea defectului în cauză, de exemplu. Același lucru este valabil și pentru un defect septal ventricular.
Corecțiile malformațiilor la extremități au o importanță secundară. După corectarea cu succes a defectului cardiac, procedurile chirurgicale reconstructive pot restabili spițele lipsă și sindromile separate. O scolioză existentă este de obicei luptată sub îngrijire fizioterapeutică. În cazuri deosebit de severe, poate fi necesară o intervenție chirurgicală pentru implantarea unei proteze de coaste de titan.
În cele mai multe cazuri, nu este necesară nicio intervenție pentru pectus carinatum. Totuși, din motive psihologice, toracele poate fi redimensionat chirurgical, urmând procedura Nuss, de exemplu.
Perspective și prognoză
Prognosticul pentru sindromul Holt-Oram este favorabil. Deși există un defect genetic, acesta poate fi tratat în mod adecvat cu opțiunile medicale actuale. Speranța de viață a cuiva cu sindrom nu este, în majoritatea cazurilor, sub media. În cazul unei malformații severe, pot exista pierderi semnificative în speranța de viață. Prognosticul este clar agravat la acești pacienți. Activitatea inimii este restricționată și poate duce la un sfârșit prematur al vieții.
Cu toate acestea, majoritatea pacienților pot fi tratate bine și cu succes. Deși nu există nicio cură din cauza defectului genetic prezent, există perspective bune pentru diverse opțiuni de corecție. Activitatea inimii este reglată și, dacă este posibil, corectată complet într-o procedură chirurgicală. Deși poate exista o afectare permanentă a modului de viață în comparație cu persoanele sănătoase, o calitate bună a vieții este obținută datorită tratamentului.
Anomaliile fizice sau malformațiile sunt schimbate într-o altă etapă. De obicei, după ce faza de creștere a copilului este completă, se inițiază o corecție necesară sau dorită a malformațiilor existente. Dacă tulburările din procesul de dezvoltare duc la tulburări semnificative, se iau măsuri corective în copilărie pentru ameliorarea simptomelor. Datorită modificărilor optice, pacientul poate avea consecințe psihologice. Acest lucru agravează prognosticul general.
profilaxie
Până în prezent, sindromul Holt-Oram nu poate fi prevenit, deoarece factorii de influență externi nu au fost clarificați în mod concludent.
Dupa ingrijire
Deoarece sindromul Holt-Oram este o boală congenitală, acesta nu poate fi vindecat complet. Prin urmare, măsurile sau posibilitățile de îngrijire ulterioară sunt foarte limitate, astfel încât persoana afectată depinde în primul rând de depistarea precoce și de tratamentul ulterior. Dacă pacientul sau părinții doresc să aibă copii, se oferă consiliere genetică pentru a preveni apariția acestui sindrom.
Tratamentul sindromului Holt-Oram are ca scop principal tratarea defectului cardiac. Acest lucru este corectat printr-o procedură chirurgicală, prin care pacientul trebuie să se recupereze și să se odihnească după procedură. Efortul sau activitatea fizică trebuie evitate. Nu este neobișnuit să fie necesar un tratament fizioterapeutic și multe dintre exerciții pot fi efectuate și în propria casă.
Cei afectați depind uneori de ajutorul și sprijinul propriei familii și prieteni. De asemenea, acest lucru poate preveni tulburările psihologice sau depresia. Mai mult, un stil de viață sănătos cu o alimentație sănătoasă are un efect foarte pozitiv asupra cursului sindromului Holt-Oram.
Puteți face asta singur
Sindromul Holt-Oram nu poate fi prevenit și, de asemenea, nu poate fi tratat cu mijloace de auto-ajutor. Cu acest sindrom, cei afectați sunt întotdeauna dependenți de o operație pentru a trata defectul cardiac pentru a prelungi speranța de viață a pacientului. Cu cât sindromul este recunoscut mai devreme, cu atât sunt mai mari șansele unui curs pozitiv al bolii. Celelalte malformații de pe corp trebuie, de asemenea, să fie corectate chirurgical.
Deoarece mulți dintre cei afectați suferă, de asemenea, de plângeri psihologice sau de complexe de inferioritate asociate cu acest sindrom, ei depind de tratamentul psihologic. Cu toate acestea, conversațiile cu alți pacienți, cu propriii părinți sau cu prietenii pot, de asemenea, să consolideze încrederea în sine a pacientului și să atenueze astfel plângerile psihologice. Mai mult, unii pacienți depind de ajutorul semenilor lor în viața de zi cu zi, în care îngrijirea caldă are un efect foarte pozitiv asupra cursului sindromului Holt-Oram.
Deoarece boala poate afecta și organele interne, pacienții sunt dependenți de examene și controale regulate de către diverși medici. Acest lucru poate preveni problemele renale, de exemplu. Copiii afectați ar trebui informați despre consecințele și complicațiile bolii.

.jpg)






.jpg)